[
вернуться к содержанию сайта]В книге рассказывается об истории развития основных направлений в квантовой химии. Авторы приводят разнообразный и малоизвестный материал, вводят читателя в круг основных понятий этой науки. Особое внимание уделяется физическому смыслу наиболее важных из них: квантовое число, валентность, молекулярная орбиталь, химическая связь и т. п. Авторы не только прослеживают историческую эволюцию системы понятий теоретической химии начиная с XIX века и до наших дней, но и показывают логическую связь между классическими и квантовыми понятиями.
Книгу с большим интересом прочтут учащиеся химических спецшкол, слушатели факультативов по химии в среднеобразовательных школах, студенты, аспиранты, интересующиеся историей химии.
За 50 лет существования квантовая химия достигла значительных успехов в объяснении физической природы химической связи, а также в определении физико-химических свойств разнообразных соединений. Круг вопросов, решаемых методами квантовой химии, в настоящее время так широк, что охватить его в небольшой книге невозможно. Поэтому авторы ограничились лишь объяснением природы химической связи и описанием её структуры.
Большое внимание в книге уделено истории возникновения и развития квантовомеханической теории химической связи. Создание этой теории было подготовлено не только открытием квантовой механики, но и всем предыдущим развитием теоретических и методологических основ химии, которые в начале XX века находились в состоянии кризиса, отразившего как кризисную ситуацию в физике того времени, так и противоречия растущей физикализации химии.
Однако тот факт, что квантовая механика является основой современного учения о химической связи, ни в коей мере не исключает огромной роли классической теории химического строения в формировании квантовохимических представлений. Действительно, теория строения в совокупности с огромным экспериментальным материалом не только стимулировала создание, но и постоянно корректировала последующее развитие теории химической связи. Хотя такое воздействие иногда трудно заметить за обилием математических формул, как показывает исследование истории вопроса, ни одно рассмотрение структурно-химических проблем не обходилось без использования в тон или иной мере эмпирических или полуэмпирических химических представлений.
Сказанное определило общую структуру книги: в гл. 1 рассмотрены доквантовомеханические модели молекул; в гл. 2 изложены некоторые концепции современной теории строения атома, причём основное внимание уделено тем, которые играли и играют существенную роль в молекулярной квантовой химии; в гл. 3 рассказано о возникновении и развитии квантовомеханической теории химической связи; гл. 4 посвящена анализу современных проблем квантовой химии, главным образом связанных с квантовомеханической интерпретацией понятий и представлений классической теории строения.
При изложении современных вопросов теории химической связи авторы использовали аппарат матрицы плотности, позволяющий наиболее просто и адекватно интерпретировать классические понятия валентности, атомного заряда и порядка связи в терминах квантовой механики.
Авторы благодарят профессора М. Г. Веселова за ценные замечания и помощь в работе.
Первые представления о химическом соединении как о целостном образовании, состоящем из противоположно заряженных частиц, находящихся в статическом равновесии, появились в начале прошлого века (Берцелиус, Дэви, Эрстед и др.). Однако в дальнейшем они не получили широкого признания. Экспериментальное открытие электрона Томсоном и Вихертом (1897 г.) создало предпосылки для появления первых электронных моделей атомов и молекул, т. е. началось выявление физического содержания понятий “химическая связь”, “валентность”, “структура” и т. п.
На развитие представлений о природе химической связи существенно повлияли физические открытия и теории, прежде всего теория атома Резерфорда — Бора. Следует отметить, что представления о природе химической связи, выдвинутые тем или иным исследователем в первой четверти XX в., существенно зависели от того, какую модель атома он принимал (или создавал). Поэтому все теории химической связи можно классифицировать по тому, какая модель [или какой тип модели: статический или динамический, резерфордовский (ядерный) или томсоновский] была положена в её основу.
Можно выделить два типа моделей химической связи.
I тип. К нему можно отнести модели, созданные до появления теории Бора (1913 г.), и модели, созданные после 1913 г., но основанные на иных, не боровских, принципах описания структуры атома (например, статическая модель атома и молекулы Льюиса).
II тип. Модели, авторы которых пытались обобщить теорию Бора на молекулярных системах, можно разделить на две группы: а) модели качественного характера (Фаулер, Сиджвик и др.); б) модели, включающие количественное описание структуры молекулярных систем, как правило, простейших (Н+2, H2),
на основе уравнений Гамильтона-Якоби с учётом условий квантования Бора-Зоммерфельда (Ниссен, Паули, Борн и др.). Эти работы фактически показали неспособность полуклассической теории типа теории Бора объяснить природу химического взаимодействия.Большинство работ качественного характера (I и II типов) связаны между собой некоторыми общими идеями, подобными тем, которые высказал Льюис в 1916 г. – обобществление электронов, идентификация валентного штриха с двумя электронами, признание фундаментальной роли правила октета.
История создания различных электронных моделей строения молекул детально описана в работах [
5, 6, 13, 25], поэтому авторы данной книги затрагивают лишь наиболее важные идеи и обращают внимание на работы, не рассмотренные ранее в историко-научной литературе.Теория ковалентной связи Льюиса
Впервые свои идеи о строении молекул американский физико-химик Джильберт Ньютон Льюис опубликовал в 1916 г., хотя первые предположения он делал гораздо раньше, ещё в 1902 г. Основное внимание учёный уделил вопросу природы химической связи в неполярных молекулах. Сопоставив полярные и неполярные соединения, чтобы выяснить, является ли разница между ними “разницей по существу или только в степени”, Льюис пришёл к выводу: “...углубляясь в обозрение всей области химических явлений, мы вынуждены... сделать заключение, что разница между наиболее полярными и неполярными молекулами есть разница только в степени и что отдельная молекула и даже часть молекул могут переходить от одного крайнего типа к другому не путём внезапного и резкого изменения, а путём неуловимых градаций” [59, с. 767]. Созданная Льюисом модель химической связи позволила ему объяснить природу указанных различий. Перейдём теперь к её рассмотрению.
Остановимся сначала на первых двух постулатах Льюиса.
1. “В каждом атоме есть некоторое ядро, остающееся неизменным при всех химических превращениях и обладающее некоторым избытком положительных зарядов, их число отвечает номеру той группы периодической таблицы, к которой относится элемент”.
2. “Атом состоит из ядра и внешней части или оболочки, содержащей отрицательные электроны, число которых в случае нейтрального атома равно избытку положительных зарядов ядра; число электронов в оболочке может изменяться при изменении химической природы атома от 0 до 8” [59, с. 768].
Таким образом, мы видим, что Льюис признаёт наличие в атоме положительно заряженного ядра. Но заряд ядра у него численно равен номеру группы Периодической системы элементов Д. И. Менделеева, где находится рассматриваемый элемент, а не порядковому номеру последнего. Поэтому и число электронов в каждом периоде изменяется, согласно Льюису, от нуля до восьми. Кроме того, в пояснениях к приведённым постулатам он указывает, что первый из них “имеет дело с двумя частями атома, которые в общих чертах соответствуют внутренним и внешним кольцам атома Томсона” [59, с. 768]. Все это говорит о том, что чёткого выбора между двумя моделями атома (резерфордовской и томсоновской) Льюисом сделано не было. Смысл, вкладываемый им в термин “ядро” (kernel), не тождествен тому смыслу, который придавали ему Резерфорд, Бор и их единомышленники, но очень близок к понятию “внутренняя часть атома”, которое использовал Томсон при обсуждении своей модели. Это хорошо видно из заключительных фраз статьи Льюиса:
“Основная трудность в изучении этих элементов (Со, Мn и др. – Прим. авт.) с помощью настоящей теории связана, как я полагаю, с тем фактом, что понятие ядра атома не является однозначно и твёрдо определённым. Вполне вероятно, что в этих элементах может происходить перенос электронов из одной части ядра в другую или между ядром и внешней оболочкой, или, возможно, между двумя отдельными внешними оболочками одного и того же атома...” [59, с. 785].
Рассмотрим теперь третий постулат. Он включает два предположения: 1) “Атом имеет тенденцию содержать чётное число электронов в оболочке” и 2) восемь электронов “располагаются симметрично в восьми углах куба” [59, с. 768].
Льюис поясняет первую мысль следующими примерами: литий имеет один электрон (на поверхности атома), фтор – семь, следовательно, электронейтральная молекула фтористого лития может быть представлена в виде LiFE8, где Е — символ электрона. Аналогично для сульфата лития можно написать Li2SO4E32, для аммиака NH3E8, для нитрата натрия NaNO3E24 и т. п. Отсюда Льюис приходит к выводу, что “...если атом имеет высшую (или низшую) степень окисления (polar number), то Е будет кратно восьми. В соединениях, в которых атомы имеют промежуточные степени окисления, число электронов не обязательно кратно восьми, но почти всегда чётное” [59, с. 770]. Например: SO2=SO2E18, NaOCl=NaOClE14 и т. п.
Те соединения, у которых Е – нечётное, “обладают высокой активностью и имеют тенденцию переходить в соединения с чётным числом электронов” [59, с. 770]. Например: NO=NOE11, NO2 = NO2E17 и т. п.
Теперь остановимся на второй части третьего постулата – идее расположения атомных электронов в углах куба. Льюис писал: “Главным соображением для принятия кубической структуры было то, что она является наиболее симметричным расположением восьми электронов, и в ней электроны наиболее удалены” [59, с. 779–780]. Иными словами, Льюис указывает на две разнородные причины принятия им кубической модели атома: соображения симметрии, которые выступают в данном случае как своеобразный эстетический принцип, и требование минимального отталкивания электронов. Однако попытки распространения кубической модели на ненасыщенные углеводороды показали, что, опираясь на эту модель, “невозможно не только представить тройную связь, но также объяснить явление свободного вращения относительно простой связи” [59, с. 780]. Это обстоятельство заставило Льюиса изменить свои первоначальные идеи, сохранив, однако, их “рациональное зерно”. В поиске новых концепций учёный обращается к началу периодической системы: “...для элементов с меньшим атомным весом, чем литий, устойчивую группу образует пара электронов – появляется вопрос, нельзя ли вообще рассматривать за основную единицу (связи — И. Д.) пару электронов, а не октет” [59, с. 779]. В дальнейшем, как известно, концепция электронной пары получила значительное развитие.
В литературе, посвящённой истории структурной химии, можно встретить мнение о том, что доквантовые электронные теории “представляли собой попытку интерпретировать простую межатомную связь как жёсткий элемент структуры, обусловленный целочисленностью валентных электронов и, по существу, исключивший вариации в энергиях связей” [16, с. 94].
Однако анализ работы Льюиса показывает, что это не совсем так. Обратимся, например, к пятому постулату: “Электроны могут с лёгкостью переходить из одного положения в наружной оболочке к другому; они удерживаются в своём положении более или менее напряжёнными
(constraints) связями, и эти положения, а также прочность связей определяются природой данного атома и тех атомов, которые соединены с ним” [59, с. 768]. Эта мысль конкретизируется в другом месте статьи Льюиса, где он рассматривает гомоатомные молекулы галогенов: “электроны, которые осуществляют связь между двумя атомами иода, удерживаются более слабыми силами, чем в случае брома и т. д., во всей группе” [59, с. 784]. Кроме того, для гетероатомных полярных молекул взаимное влияние атомов обусловлено по Льюису различным по величине притяжением электронной пары, осуществляющей химическую связь, к разным атомам, что выражается в различной полярности соединений. Рассматривая молекулу H2ClC–COOH, Льюис говорит о постепенном ослаблении “разделения электронов между атомами при удалении от атома хлора” [59, с. 782]. С помощью идей Льюиса многие американские и английские химики разработали в 20-х годах электронные модели взаимного влияния атомов. Так, например, в 1923 г. ученики Льюиса Латимер и Родебуш исследовали способность электроотрицательного атома изменять свойства соседних функциональных групп.Как сказано в работе [
16], рациональный химический динамизм связан не с механическим взглядом на атомы и молекулы, а с появлением представлений о неравноценности сил и энергий химической связи и вообще химического взаимодействия, в том числе взаимного влияния атомов [16, с. 93]. Поэтому нужно отметить, что теория Льюиса вовсе не исключает указанные идеи, и когда мы говорили о ней как о статической теории, это не следует понимать буквально – она статична по сравнению с теорией Н. Бора, но не в смысле “рационального химического динамизма”.С квантовохимической точки зрения понятно, почему гипотеза о статическом атоме (при отсутствии в нём орбитального движения электронов) в совокупности с предположением о взаимной проницаемости атомных оболочек (четвёртый постулат) дала возможность качественно рассмотреть ковалентную связь. Действительно, согласно теореме Гельмана–Фейнмана, распределение электронной плотности в молекуле, определяемое одночастичной матрицей плотности ρ, таково, что силы, действующие на ядра, могут быть рассчитаны по законам классической электростатики суммированием вкладов от каждого элемента статического объёмного заряда, “размазанного” в пространстве с плотностью ρ. Это обусловило впоследствии успех многих квантовохимических методов, особенно тех из них, в которых развивается квазиклассический подход к определению типа ядерного полиэдра молекулы, например модель Сиджвика и Пауэлла, развитая затем Гиллеспи и Найхолмом (подробнее см. [
9]).В 20-х годах была дана качественная трактовка реакций присоединения к насыщенным молекулам, структуры ряда комплексных соединений, а также в первом приближении объяснена природа водородной связи. Это удалось сделать с помощью выдвинутой Льюисом и развитой впоследствии Сиджвиком [
78, 79] концепции неподелённой (свободной) электронной пары, способной образовывать химические связи.Значение появления этой концепции трудно переоценить. Если в конце XIX – начале XX вв. для объяснения существования многих комплексных соединений и протекания реакций присоединения к насыщенным молекулам приходилось прибегать к искусственным представлениям о “дополнительных” (скрытых, побочных) валентностях, то с появлением модели Льюиса и концепции неподелённых электронных пар необходимость в подобных построениях отпала. По словам Сиджвика: “обе ветви химии – органическая и неорганическая — получили благодаря введению электронных представлений единый теоретический фундамент” [
79, с. 468].Большая заслуга в разработке и пропаганде идей Льюиса принадлежит американскому физикохимику Ирвингу Ленгмюру. По образному замечанию американского историка химии М. Зальцмана: “если бы не Ленгмюр, то ключ к химической связи оказался бы надолго зарытым в химической литературе” [77].
Основные идеи своей работы [
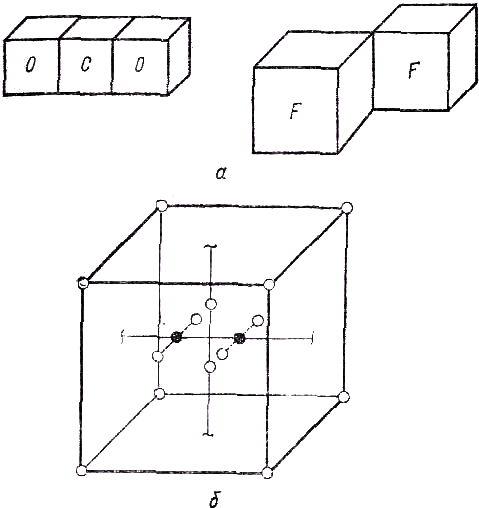
57] Ленгмюр выразил в одиннадцати постулатах, большая часть которых относится к строению электронной оболочки. Модель Ленгмюра, так же как и модель Льюиса, – электростатическая. Оба автора пытаются связать её с ранней моделью Томсона. Но у теории Ленгмюра имеются некоторые преимущества, главное из которых – принцип заполнения электронных оболочек, которые Ленгмюр разбивает на “ячейки” (cells). В каждой ячейке может находиться не более двух электронов*. Следует заметить, что этот принцип заполнения электронных оболочек был распространён Ленгмюром на все известные тогда химические элементы. Но главное, что интересно в данной книге, – это его взгляд на природу химической связи. Ленгмюр выделяет два типа стабильных электронных конфигураций: электронную пару и октет. При образовании химической связи все валентные электроны участвуют в образовании октетов, либо переходя от одного атома к другому, либо путём образования общих электронных пар. Общее число электронов е, число октетов п и число электронных пар р, “удерживаемых сообща (hold... in common) октетами”, Ленгмюр связал формулой 2р=8n–е.На рис. 1 показаны некоторые схемы электронного строения молекул, взятые из работы [
57].Особого внимания заслуживает десятый постулат Ленгмюра, точнее, его вторая часть: “В исключительных случаях октет может образовываться около сложного ядра, т. е. около структуры, содержащей ядра двух атомов, удерживаемых вместе парой электронов” [
57, с. 888].Примером такого “исключительного случая” является молекула азота. Необычайная стабильность и химическая инертность этой молекулы была объяснена Ленгмюром тем, что она имеет следующее электронное строение: каждое ядро атома удерживает пару электронов первой оболочки (т. е., говоря современным языком, 1
s-электроны не принимают участия в химической связи); восемь из оставшихся десяти электронов образуют октет (см. рис. 1, б), внутри которого, между ядрами азота, находятся два электрона.
Рис. 1. Электронные модели молекул по Ленгмюру:
а — молекулы СО
2 и F2; б — молекула N2В этой же работе Ленгмюр впервые сформулировал принцип изоэлектронности (по его терминологии “изостерности”). В качестве одного из примеров изоэлектронных серий был рассмотрен ряд молекул: N2, СО и CN–. Способ описания молекулы СО и аниона
CN– такой же, как и молекулы N2. В указанном отрывке из 10-го постулата и приведённых примеров можно видеть начало принципиально нового способа описания молекул, которому в квантовой химии соответствует метод молекулярных орбиталей. Разумеется, речь идёт не о детальном сходстве, а об аналогии в самой постановке задачи изучения электронной структуры молекулы. Суть этой аналогии заключается в том, что и в методе молекулярных орбиталей (МО), и в отдельных построениях Ленгмюра молекула рассматривается как “многоядерный атом”, т. е. допускается, что при решении молекулярной задачи можно применить принципы, подобные тем, которые используются в теории атома при анализе заполнения электронных оболочек атомов элементов.Чтобы пояснить эту мысль, обратимся снова к работе Ленгмюра. Наиболее стабильная восьмиэлектронная оболочка атомов инертных газов представлялась ему, как и Льюису, кубом, в вершинах которого находятся электроны. С другой стороны, Ленгмюр в отличие от Льюиса рассматривает и такие модели молекул (они указаны выше), в которых последние “устроены” наподобие атомов инертных газов, т. е. оба ядра и внутренние электроны окружены октетом электронов, аналогичным октету электронов второй
(L) оболочки атома.В методе МО предполагается, что электроны находятся на орбиталях, охватывающих все ядра в молекуле. Электроны молекулы распределяются при этом по молекулярным орбиталям с учётом принципа минимума энергии и ограничений, налагаемых запретом Паули, что аналогично принципу построения электронных оболочек в теории атома.
При использовании изоэлектронного принципа Ленгмюра в тех случаях, когда речь шла о молекулах с общим октетом электронов, возникали некоторые трудности. Так, молекулы
CN, СО и N2, NO должны иметь соответственно один, два и три валентных электрона, не принадлежащих к первой (K) оболочке, но заключённых внутри октета. Как заметил Малликен: “...с этой точки зрения, эти молекулы должны быть подобны атомам Na, Mg и А1. Однако никакой аналогии в их химическом поведении не видно. По химическим свойствам молекула CN похожа скорее на С1, чем на Na, (...), a N2 — на аргон, чем на Mg” [65, с. 188].Однако в 1925 г. Малликен обратил внимание на сходство электронного спектра молекулы CN и ряда других изоэлектронных ей систем с одним валентным электроном в октете (СО+, N2+, ВО и др.) со спектром Na “в отношении природы и расположения электронных уровней”.
В 1926 г. Бердж показал, что электронные уровни молекул СО и N2 аналогичны уровням Mg. Подмеченные аналогии послужили затем толчком к созданию метода МО (см. гл. 3).
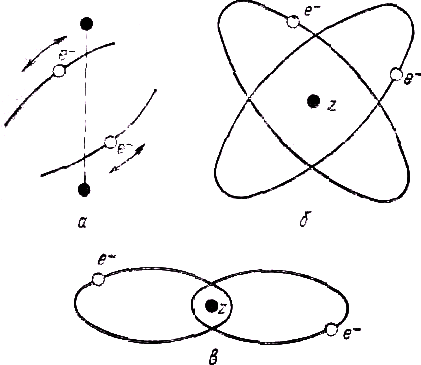
Рис. 2. Динамические модели молекулы водорода:
а — по Ленгмюру; б — по Бору-Кэмблу-Паули (модель “перекрещивающихся орбит”); в — по Зоммерфельду-Гейзенбергу
Таким образом, при сопоставлении идей Льюиса (1916 г.) с идеями Ленгмюра (1919 г.) можно прийти к следующему выводу: ещё до возникновения квантовой химии в недрах электронных теорий наметились два подхода к изучению электронного строения молекулы. В рамках одного из них связь между атомами осуществляется общей электронной парой (Льюис), что нашло затем своё квантовомеханическое отражение в приближении идеального спаривания метода валентных связей (ВС). Другой подход (Ленгмюр) допускает обобществление большего числа электронов, при этом на молекулы переносится принцип, аналогичный принципу заполнения электронных оболочек атома, что, будучи переведённым на современный язык, соответствует основной идее метода МО
**.* Исключение составляют только две ячейки первой оболочки, они могут вмещать только по одному электрону каждая.
** Оба указанных выше подхода имеют глубокие исторические корни. Ещё в XIX в. сложились два взгляда на строение молекул – структурный (А. М. Бутлеров, Кекуле и др.) и унитарный (Бодримон, Лоран, Жерар, Н.А. Меншуткин и др.), что обусловлено неоднозначностью реализации дальтоновского представления о молекуле как системе взаимодействующих атомов. Оба подхода получили дальнейшее развитие как в ранних электронных теориях, так и в квантовой химии.
СПИСОК ЛИТЕРАТУРЫ
1. Алексеев В. Г.
2. Берсукер И. Б. Электронное строение и свойства координационных соединений. Изд. 2-е. Л., “Химия”, 1976.
3. Борисова Н. П., Семёнов С. Г. Молекулярно-орбитальное определение кратности химической связи. — “Вестн. Ленингр. ун-та”, 1973, № 16, с. 119—124.
4. Борисова Н. П., Семёнов С. Г. Молекулярно-орбитальное исследование валентной структуры органических соединений. — “Вестн. Ленингр. ун-та”, 1976, № 16, с. 98—103.
5. Быков Г. В. История электронных теорий органической химии. М., Изд-во АН СССР, 1963.
6. Быков Г. В. История органической химии. М., “Химия” 1976, 360 с.
7. Вейль Г. Валентность и иерархия химических структур. – В кн.: Прикладная комбинаторная математика. Пер. с англ. Под ред. М. Е. Деза. М., “Мир”, 1968, с. 339—350.
8. Вигнер Е. Теория групп и её приложения в квантовомеханической теории атомных спектров. Пер. с англ. Под ред. И. Я. Смородинского. М., Изд-во иностр. лит., 1961.
9. Гиллеспи Р. Геометрия молекул. Пер. в англ. Под ред. Ю. А. Пентина. М., “Мир”, 1975.
10. Горьков Л. П., Питаевский Л. П. Энергия расщепления термов молекулы водорода. — “Докл. АН СССР”, 1963, т. 151, с. 822—825.
11. Дмитриев И.С. Симметрия в мире молекул. Л., “Химия”, 1976.
12. Дмитриев И. С. Молекулы без химических связей. Очерки о химической топологии. Л., “Химия”, 1979.
13. Дмитриев И. С. Развитие электронных теорий ковалентной связи в работах Дж. Льюиса и И. Лэнгмюра. — В кн.: Вопросы истории и методологии химии. Под ред. Р. Б. Добротина. Л., Изд-во ЛГУ, 1976, с. 98—116.
14. Каплан И. Г. Симметрия многоэлектронных систем. М., “Наука”, 1969.
15. Клейн Ф. Сравнительное обозрение новейших геометрических исследований (Эрлангенская программа). Пер. с нем. — В кн.: Об основаниях геометрии. Под ред. А. П. Нордена. М., Гостехиздат, 1956, с. 399—451.
16. Кузнецов В. И. Диалектика развития химии. М., “Наука”, 1973, 303 с.
17. Местечкин М. М. Метод матрицы плотности в теории молекул. Киев, “Наукова думка”, 1977.
18. Пальм В. А. Введение в теоретическую органическую химию. М., “Высшая школа”, 1974.
19. Полинг Л. Химики —это те, кто на самом деле понимают мир. — “Химия и жизнь”, 1976, № 2, с. 92–97.
20. Родный Н. И. Общая характеристика состояния науки периода 1800—1870 гг. — В кн.: Проблемы развития науки в трудах естествоиспытателей XIX века. Под ред. Б. С. Грязнова и др. М., “Наука”, 1973, с. 7-28.
21. Рюденберг К. Физическая природа химической связи. Пер. с англ. Под ред. А. М. Бродского. М., “Мир”, 1964.
22. Семёнов С. Г. Локализация молекулярных орбиталей методом эталонной матрицы плотности. — “Вестн. Ленингр. ун-та”, 1978, № 10, с. 107—113.
23. Семёнов С. Г. Анализ заселённостей и проблема неортогональности многоцентрового базиса атомных орбиталей. — Там же, 1977, №22, с. 125—131.
24. Сизова О. В., Барановский В. И. Электронное строение, донорно-акцепторные свойства и взаимное влияние лигандов в комплексах [PtX3L]n. — “Теор. и эксперим. химия”, 1975, т. 11, с. 147—155.
25. Соловьёв Ю. И. История химии. Т. 1. М., “Просвещение”, 1977.
26. Фок В.А. О волновых функциях многоэлектронных систем.— “Журн. эксперим. и теор. физ.”, 1940, т. 10, вып. 9—10, с. 967—979.
27
. Чаркин О. П., Бобыкина Г. В., Дяткина М. Е. Орбитальные потенциалы ионизации атомов и ионов в валентных конфигурациях. - В кн.: Строение молекул и квантовая химия. Киев, “Наукова думка”, 1970, с. 155—175.28. Armstrong D. R., Fortune R., Perkins P. G.
A molecular orbital study of the trans effect. — “Inorg. chim. Acta”, 1974, v. 9, p. 9—18.29. Armstrong D. R., Perkins P. G., Stewart J. J. P. Bond indices and valency. — “J. Chem. Soc. Dalton Trans.”, 1973, p. 838— 440.
30. Armstrong D. R., Perkins P. G., Stewart J. J. P. Valencies and bond indices for the elements from hydrogen to clorine. — “J. Chem. Soc. Dalton Trans.”, 1973, p. 2273—2277.
31. Boys S. F. Localized orbital” and localized adjustment functions. — In: Quantum theory of atoms, molecules, and the solid state. N. Y., 1966, p. 253—262.
32. Cayley A. On the mathematical theory of isomers. — “Phil. Mag”, 1874, v. 47, p. 444–449.
33. Christoffersen R. Е., Baker К. A.
Electron population analysis. Gross atomic charges in molecules.— “Chem. Phys. Lett.”, 1971, v. 8, p. 4—9.34. Clifford W. Letter to J. J. Sylvester. — “Amer. J. Math.” 1878, v. 1, p. 126—128.
35. Clinton W. L., Nakhleh J., Wunderlich F. Direct determination of pure-state density matrices—“Phys. Rev.”, 1969, v. 177, p.1.
36. Coulson С. A
., Moffit W. E. The properties of certain strained hydrocarbons. — “Phil. Mag.”, 1949, v. 40, p. 1—35.37. Davidson E. R. Electronic population analysis of molecular wave functions. — “J. Chem. Phys.”, 1967, v. 46, p. 3320.
38. Edmiston С., Ruedenberg К.
Localized atomic and molecular orbitals. — “J. Chem. Phys.”, 1965, v. 43s, p. 97—115.39. Fock V. Naherungsmethode zur Losung des quantenmechanischen Mehrkorperproblems. — “Z. Phys.”, 1930, Bd 61, S. 126—148.
40. Galiup G. A., Norbeck J. M. Population analysis of valencebond wave
functions and ВеН2. — “Chem. Phys. Lett.”, 1973 v. 21, p. 495—500.41. Gaunt J. A. Theory of Hartree's atomic fields. — “Proc. Cambridge Phil. Soc.”, 1928, v. 24, p. 328—342.
42. Giambiagi M., Giambiagi M., Grempel D. R. e. a. Sur la definition d'um indice de liaison (TEV) pour des bases non orthogonales. Proprietes et applications. — “J. Chim. Phys.”, 1975, v. 72, p. 15—22.
43. Goddard W. A. Improved quantum theory of many-electron systems. — “Phys. Rev.”, 1967, v. 157, p. 73—93.
44. Goddard W. A., Wilson C. Exchange kinetic energy contragradience and chemical binding. — “Chem. Phys. Lett.”, 1970, v. 5, p. 45—49.
45. Gordan P., Alexeiew W. Ubereinstimmung der Formeln der Chemie und der Invariantentheorie. — “Z. Phys. Chem.”, 1900, Bd 35, H. 5, S. 610—633.
46. Grabenstetter J. E., Whitehead M. A. CNDO: Problems in electron population analysis. — “J. Chem. Soc. Faraday Trans.”, 1973, N 7, p. 962—967.
47. Hartree D. R. The wave mechanics of an atom with a non-coulomb central field. — “Proc. Cambridge Phil. Soc.”, 1928, v. 24, p. 89—110.
48. Heitler W. Gruppentheorie der homoopolaren chemischen Bindung. — “Z. Phys.”, 1928, Bd. 47, S. 835—849.
49. Heitler W. Quantentheorie der Valenz. — “Naturwiss.”, 1929, Bd 17, S. 546—574.
50. Heitler W., London F. Wechselwirkung neutral Atome und homoopolare Bindung nach der Quantenmechanik. — “Z. Phys.”, 1927, Bd 44, S. 455—472.
51. Heitler W., Rumer G. Quantenchemie mehratomiger Molekule. — “Nachr. Gott. Math.-Physib, 1930, H. 3, S. 277.
52. Heitler W., Rumer G. Quantentheorie der chemischen Bindung fur mehratomige Molekule. — “Z. Phys.”, 1931, Bd 68, S. 12.
53. Herzberg G. Zur Bildung der Zweiatomige Molekule. — “Z. Phys.”, 1929, Bd 57, S. 601–612.
54. Hund F. Zur Deutung der Molekulspektren. — “Z. Phys.”, 1927, Bd 40, S. 742—764; Bd 42, S. 93–120; Bd 43, S. 805—826; 1928; Bd 51, S. 759–795; 1930, Bd 63, S. 719.
55. Hund F. Molekulbildung. — “Ergeb. exact. Naturwiss.”, 1929, Bd 8, S. 147–201.
56. Jammer M. The conceptual development of quantum mechanics. N. Y., McGraw-Hill Book Company, 1966, 356 p.
57. Langmuir I. The arrangement of electrons in atom and molecules. — “J. Amer. Chem. Soc.”, 1919, v. 41, p. 868–934.
58. Lennard-Jones J. E. The electronic structure of some diatomic molecules. — “Trans. Farad. Soc.”, 1929, v. 25, p. 668.
59. Lewis G. N. The Atom and the Molecule. — “J. Amer. Chem. Soc.”, 1916, v. 38, p. 762—785.
60. London F. Quantentheorie der homoopolaren Valenzzahlen. — “Z. Phys.”, 1928, Bd 46, S. 455—478.
61. London F. Quantenmechanik der homoopolaren Valenzchemie. — “Z. Phys.”, 1928, Bd 50, S. 24-52.
62. Lowdin P.-O. On the nonorthogonality problem. — “Adv. Quant. Chem.”, 1970, v. 5, p. 185—199.
63. McWeeny R., Del Re G. Criteria for bond orbitals and optimum hybrids. — “Theor. Chim. Acta”, 1968, v. 10, p. 13—22.
64. Mulliken R. S. Class of one-valence-electron emitters of band spectra. — “Phys. Rev.”, 1925, v. 26, p. 561—573.
65. Mulliken R. S. The assignment of quantum numbers for electrons in molecules. — “Phys. Rev.”, 1928, v. 32, p. 186— 222, 761—814.
66. Mulliken R. S. Electronic structures of polyatomic molecules and valence. — “Phys. Rev.”, 1932, v. 41, p. 49—71.
67. Mulliken R. S. Electronic population analysis on LCAO-MO molecular wave functions. — “J. Chem. Phys.”, 1955, v. 23, p. 1833—1840.
68. Martensson O., Ohrn Y. Complex hybridization of orbitals of s- and p-types.— “Theor. Chim. Acta”, 1967, v. 9, p. 133—139.
69. Matsen F. Chemistry without spin. — “J. Amer. Chem. Soc.”, 1970, v. 92, p. 3525—3538.
70. Offenhartz P. SCF wavefunctions and energies for valencestates and excited of carbon and nitrogen. — “Chem. Phys. Lett.”, 1973, v. 21, p. 388—394.
71. Pauling L. The shared-electron chemical bond. — “Proc. Nat. Acad. Sci.”, 1928, v. 14, p. 359—362.
72. Pauling L. The nature of the chemical bond. — “J. Amer. Chem.Soc.”,1931, v. 53, p. 1367—1400, 1932, v. 54, p. 988.
73. Polak R. Optimum hybrid orbitals in localized orbitals. — “Int. J. Quant. Chem.”, 1970, v. 4, p. 271—287; 1972, v. 6, p. 1077—1086.
74. Roby К. R.
Quantum theory of chemical valence concepts. — “Mol. Phys.”, 1974, v. 27, p. 81—104; v. 28, р. 1441—1456.75. Roothaan С. С. J.
New developments in molecular orbital theory. — “Rev. mod. Phys.”, 1951, v. 23, p. 69—89.76. Rumer G. Zur theorie der Spinvalenz. — “Nachr. Gott. Math.-Phys.”, 1932, H. 4, S. 337—341.
77. Saltzman M. D. Benzene and the triumph of the octet theory.— “J. Chem. Educ.”, 1974, v. 51, p. 498—502.
78. Sidgwick N. V. The electronic theory of valence. Oxford, Univ. Press, 1927, 223 p.
79. Sidgwick N. V. The nature of the non-polar link. — “Trans. Faraday Soc.”, 1923, v. 19, p. 468—475.
80. Slater J. C. The theory of complex spectra, — “Phys. Rev.”, 1929, v. 34, p. 1293—1323.
81. Sylvester J. J. On an application of the new atomic theory. — “Amer. J. Math.”, 1878, v. 1, p. 64—104.
82. Switkes E., Stevens R. M., Lipscomb W. N. e. a. Localized bonds in SCF wave functions for polyatomic molecules. — “J. Chem. Phys.”, 1969, v. 51, p. 2085—2093.
83. Waller I., Hartree D. R. On the intensity of total scattering of X-rays. — “Proc. Roy. Soc.”, Ser. A., 1929, v. 124, p. 119.
84. Walsh A. D. The.structures of ethylene oxide, ciclopropane and related molecules. — “Trans. Faraday Soc.”, 1949, v. 45, p. 179—190.
85. Weyl H. Gruppentheorieund Quantenmechanik, Leipzig, 1928, S. 328.
86. Weyl H. Zur quantentheoretischen Berechnung molecular Bindungsenergien. — “Nachr. Gott. Math.-Phys.”, 1930, H. 3.
87. Weyl H., Teller E., Rumer G. Eine fur die Valenztheorie geeignete Basis der binaren Vektorinvarianten, — “Nachr. Gott. Math.-Phys.”, 1932, Hf. 6, S. 499—504.
88. Wiberg
К. В. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinol and cyclobutilcation and to bicyclobutane. — “Tetrahedron”, 1968, v. 24, p. 1083.89. Wiberg К. В., EllisonG. В., Wendoloski J. J.
Electronic states of organic molecules. — “J. Amer. Chem. Soc.”, 1976, v. 98, p. 1212—1218.Дата установки:
24.02.2008
![]()
![]()